
1. Fastqファイルの前処理とは
ダウンロードしてきた(もしくは自分で取得した)Fastqファイルは、いわば不純物を含んだ「原石」の状態で、そのまま解析に使うのは好ましくありません。
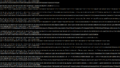
@DRR212438.35 35 length=101
CTTGACAGGAAGTCTGGTTGGTACAGCAAAAGAAATGTGATGATATTTGATTTAGGTGGTGGTACTTTAGATGTTTCACTACTTACCATAGCTGATAGCGT
+DRR212438.35 35 length=101
<AFFFFJJJJJJ7AJFF-FJJFJFAJJ-AFFJJAFJF7<<FF7FJJFFA-<AFFJ--F-FA<FFJFJJJ<AFAJFAJFAAFJ<-<F<FFFFF-AF7A<<F-例えば上記のリードでは、左から17塩基目まではクオリティスコア20を超えていて問題ありませんが、18塩基目以降は「-」(クオリティスコア12)が出現するなど、あまり信頼できない配列が目立ちます。このように、信頼度の低い塩基は解析から除外しておく必要があります。
また、Illuminaによるシークエンスでは、「アダプター配列」と呼ばれる、DNA断片をフローセルに結合させたり、シークエンシングプライマーを結合させたり、複数サンプルを同時に解析したりする際のバーコード(目印配列)の役割をする配列をライブラリ調製の段階で結合させており、それがFastqファイルにも残っている場合があります。このようなアダプター配列も解析の邪魔になりますので、除去する必要があります。
2. Fastqファイルのトリミングツール
Trimmomaticが有名です。これ一つでアダプターの除去からリードのトリミングまでを実行することができます。オプションが豊富で、トリミング条件を細かく設定することができます。しかし、慣れない頃はこれが逆に悩みの種になってしまったりします。特に、アダプタートリミングを行うためにアダプター配列を指定しないといけないところは初心者泣かせです。そこで、本稿ではPEATを用いてアダプタートリミングを行い、その他のクオリティトリミングをTrimmomaticで行う方法を紹介します。
2.1 PEATのダウンロード
PEATの特徴は何といっても「アダプター配列を自動で検出してくれること」です。これによりFastqファイルの作成者(実験者)がライブラリ調製時に用いたアダプターの種類が不明であっても、頻出する配列を自動で探し出してトリミングを行ってくれます。
上記のgithubサイトからコンパイル済のバイナリファイルをダウンロードできます。v1.2.4が最新版のようです(2022年9月3日現在)。
#Linux用
$wget https://github.com/jhhung/PEAT/releases/download/v1.2.4/PEAT_linux_v1.2.4
#Mac用
$curl https://github.com/jhhung/PEAT/releases/download/v1.2.4/PEAT_mac_v1.2.42.2 PEATの使い方
Linux版の使用法を例に示します。PEATのパスとfilenameの部分を自身のものに変更してください。
$PEAT_linux_v1.2.4 \
paired \ #ペアエンドの場合(シングルエンドの場合はsingle)
-1 filename_1.fq.gz \ #Fastqファイルはgzip圧縮していても可
-2 filename_2.fq.gz \
-o filename \ #トリミング済Fastqのファイル名を指定
-n 2 \ #解析に使用するCPUコア数(PCの能力以上を指定した場合は自動調整)
--adapter_min_bp 10 \ #アダプターとして検出する最小の塩基数
--adapter_contexts \ #検出されたアダプター配列を10位まで表示
--verbose #解析の進捗をリアルタイムで表示アダプタートリミング済のFastqファイルは、「filename_paired1.fq」および「filename_paired2.fq」という名前で出力されます。これらを更にTrimmomaticを用いたクオリティトリミングにかけていきます。
2.3 Trimmomaticのダウンロード

上記の配布サイトからVersion 0.39(2022年9月3日時点で最新)のbinary版をダウンロードします。
$wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.39.zip
$unzip Trimmomatic-0.39.zip
$cd Trimmomatic-0.39
$ls
LICENSE adapters trimmomatic-0.39.jar2.4 Trimmomaticの使い方
一例を示します。トリミング条件の意味も合わせて示しますので、慣れてきたらいろいろと条件検討してみて下さい。
$java -jar trimmomatic-0.39.jar \
PE \ #ペアエンドの場合
-threads 2 \ #使用CPUコア数
-trimlog filename_trimmolog.txt \ #ログファイル名
filename_paired1.fq \ #1つ目のFastq(アダプタートリミング済)
filename_paired2.fq \ #2つ目のFastq(アダプタートリミング済)
filename_cleaned_1.fq \ #1つ目のFastqのクオリティトリミング済出力(paired)
filename_unpaired_1.fq \ #1つ目のFastqのクオリティトリミング済み出力(unpaired)
filename_cleaned_2.fq \ #2つ目のFastqのクオリティトリミング済出力(paired)
filename_unpaired_2.fq \ #2つ目のFastqのクオリティトリミング済出力(unpaired)
LEADING:20 \ #リードの先頭からクオリティスコアが20未満の塩基を除去
TRAILING:20 \ #リードの後尾からクオリティスコアが20未満の塩基を除去
SLIDINGWINDOW:10:20 \ #10塩基毎に平均クオリティスコアを計算し、20未満であればそれ以降のリードを除去
MINLEN:20 #20塩基未満のリードを除去「filename_cleaned_1.fq」および「filename_cleaned_2.fq」がトリミング済のfastqファイルのペアになります。
3. References
Anthony M. Bolger, Marc Lohse, Bjoern Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, Volume 30, Issue 15, 1 August 2014, Pages 2114–2120, https://doi.org/10.1093/bioinformatics/btu170
Li, YL., Weng, JC., Hsiao, CC. et al. PEAT: an intelligent and efficient paired-end sequencing adapter trimming algorithm. BMC Bioinformatics 16 (Suppl 1), S2 (2015). https://doi.org/10.1186/1471-2105-16-S1-S2
Follow me!

コメント