Illuminaなどのシーケンサーで取得した塩基配列は、「FASTQ形式」で記述されています。すべての解析は、このFastqファイルを入手するところから始まります。ここでは、インターネット上のデータベース上に公開されているFastqファイルをダウンロードする手順についてご紹介します。

1. Fastqファイルとは?
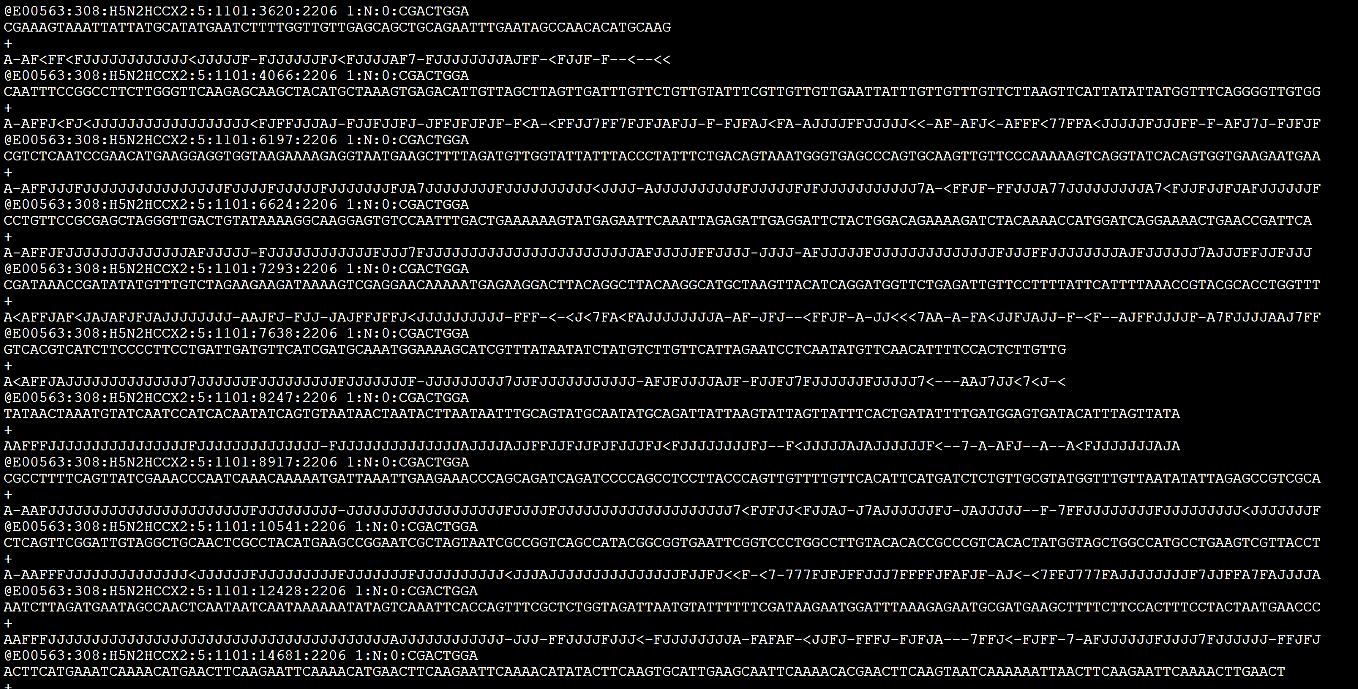
「xxxxx.fastq」や、「xxxxx.fastq.gz」(xxxxxは任意の名前)というファイル名で保存されており、塩基配列とそのクオリティが記述されたテキストファイルのことです。試しに開いてみると、下のような文字列が表示されます。
※デスクトップに保存したFastqファイルをダブルクリックで開かないように。大きなものでは数GBもありますので、一発でPCがフリーズします。開くときはターミナルを起動して、「less」(gzファイルの場合は「zless」)コマンドを使用して開くようにしましょう。
less DRR212461_1.fq
@DRR212461.4 4 length=101
CTTCCAACCCCAGCCCTTCTCCGCCTCAGTCCGCCGCCATAGTCGCCGGACAAAACACAAGCAGGGGAGCCAAGTGACCATGTCAGCTTCATTGAGTTTTG
+DRR212461.4 4 length=101
AAFFFJJJJJJJJJJJJJJJJFJJJJJ<FFJJJJJJJJJJJJJJAJJJJJFJJJJJJJJJJJJJJJJJJJJJJJFJJFJJJJJJJJJJFJJAJJJJA<AJご覧のとおり、1本の配列(リード)は4行で記述されています。1行目は@で始まっており、配列名(ヘッダー)を表しています。2行目には塩基配列が記されています。3行目は+で始まっており、再びヘッダー行(ヘッダー名がない場合もあります)、4行目には2行目の塩基配列に対応するクオリティ値(Phredクオリティスコア)が記載されています。2行目と4行目は同じ文字数になっています。これが延々と繰り返され、1つのFastqファイルが構成されているわけです。
クオリティ値(Phred Quality Score: Q)は、
Q = -10log10(p) ⇔ p = 10(-Q/10)
で表されます。Q値は大きいほどそのベースコールが確からしいことを示します。例えばQ = 10のとき、p = 0.1となりますが、これは、0.1の確率でそのベースコールは正しくない、つまり0.9の確率で正しいことを表します。同じ理屈で、Q = 20であれば、そのベースコールは0.99の確率で正しいことを示し、通常NGS解析では、Q > 20の基準で塩基配列のトリミングを行うことが多いです。
なお、Fastqで表示されているクオリティ値(英数字)はPhred+33という形式で記述されており、その英数字のASCIIコードを調べて33を引くとQ値が求められるようになっています(詳しく知りたい方はこちらを参照)。例えば上記のFastqの配列の一文字目「C」のクオリティ値は「A」となっています。「A」のASCIIコードは65ですので、Q = 65 – 33 = 32となります。

2. Fastqファイルの探し方
Fastqファイルをダウンロードするにあたり必要な情報は、「SRA Run accession No.」です。ERRxxxxxxやDRRxxxxxxのような9桁の番号で、シークエンスデータに直接紐づいています。SRA(Sequence Read Archive)の仕組みについて詳しく知りたい方はこちら。
きちんとした雑誌に発表された論文であれば、使用したシークエンスデータは必ずWeb上で公開されています(そうでないと査読をクリアできません)。さらに、親切な文献であれば、Supplementary materials等にSRA Run accession No.を示してくれていることもあります(管理者もそうしています)。しかし、そうでない不親切な論文も多く、目的のシークエンスデータになかなかたどり着けない経験をされた方も多いのではないでしょうか?
そこで以下に管理者の探索手順を示しますので、参考になれば幸いです。

2.1. Bioproject accession numberを入手する
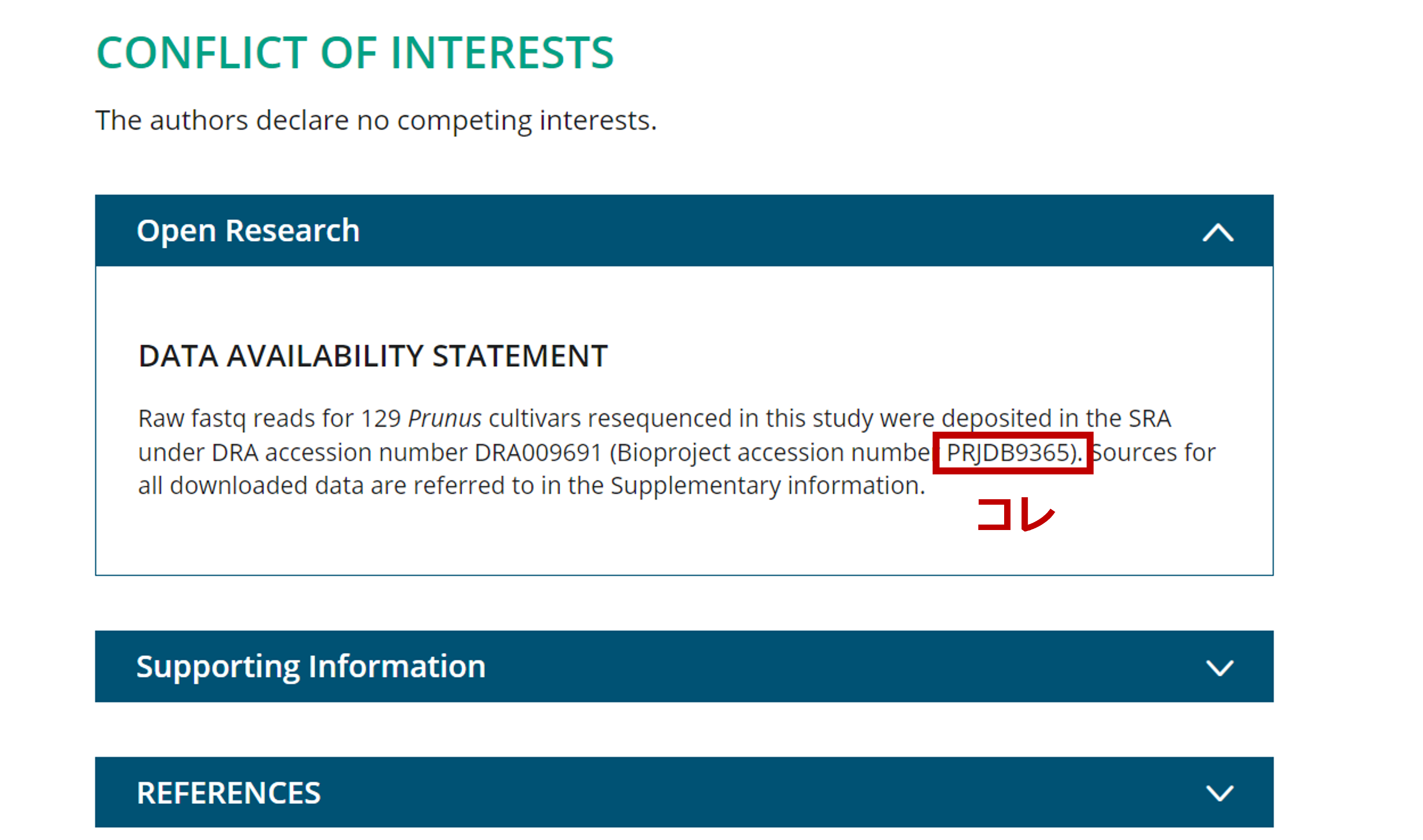
論文には大抵 Data availability statementというセクションがあり、そこにPRJDBxxxxのようなアクセッション番号が記載されています(どんなに不親切な論文でも、流石にこれくらいの記載はあります)。
Numaguchi et al. (2020)の例
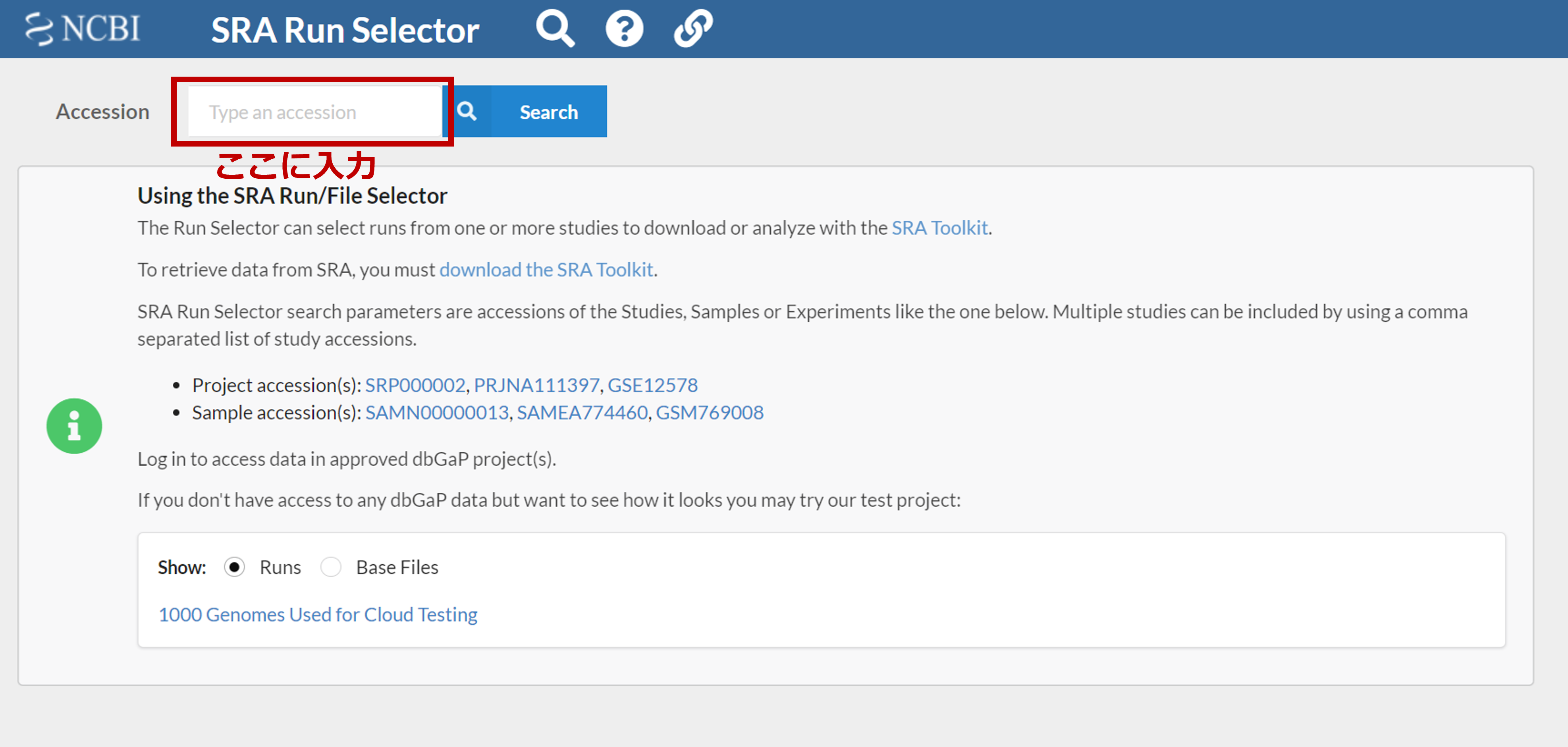
2.2 SRA Run SelectorにBioproject accession numberを入力し、データを検索する。

すると、下記のような検索結果が表示されます。
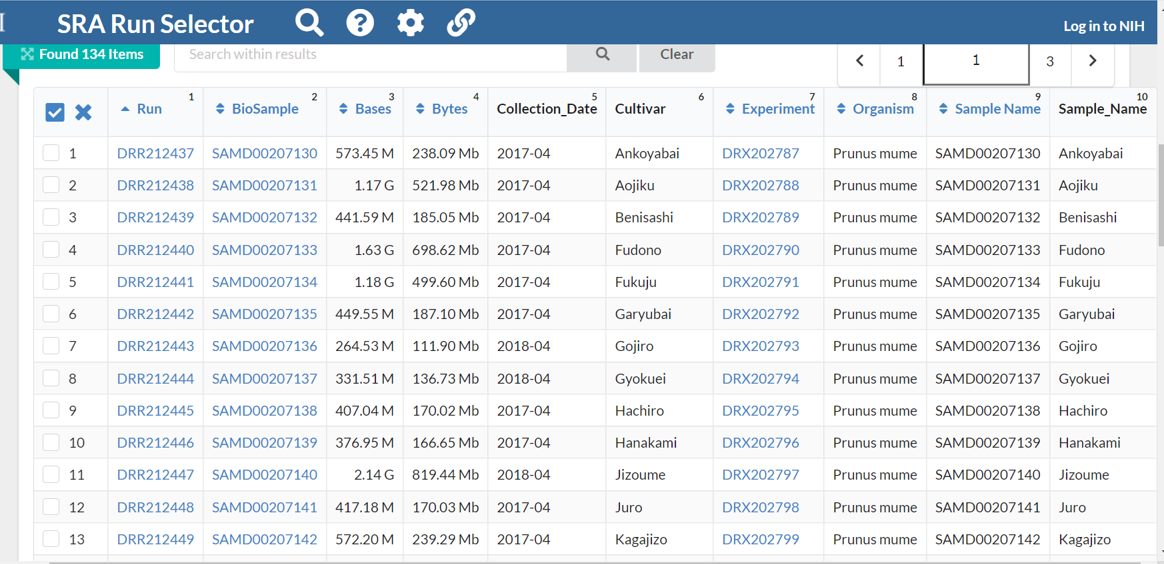
このうち、「Run」列に表示されているのが目的のSRA Run accession No.です。これらに対応するサンプル(品種・系統)名が「Cultivar」列に表示されています(ただし、これは親切な例)。
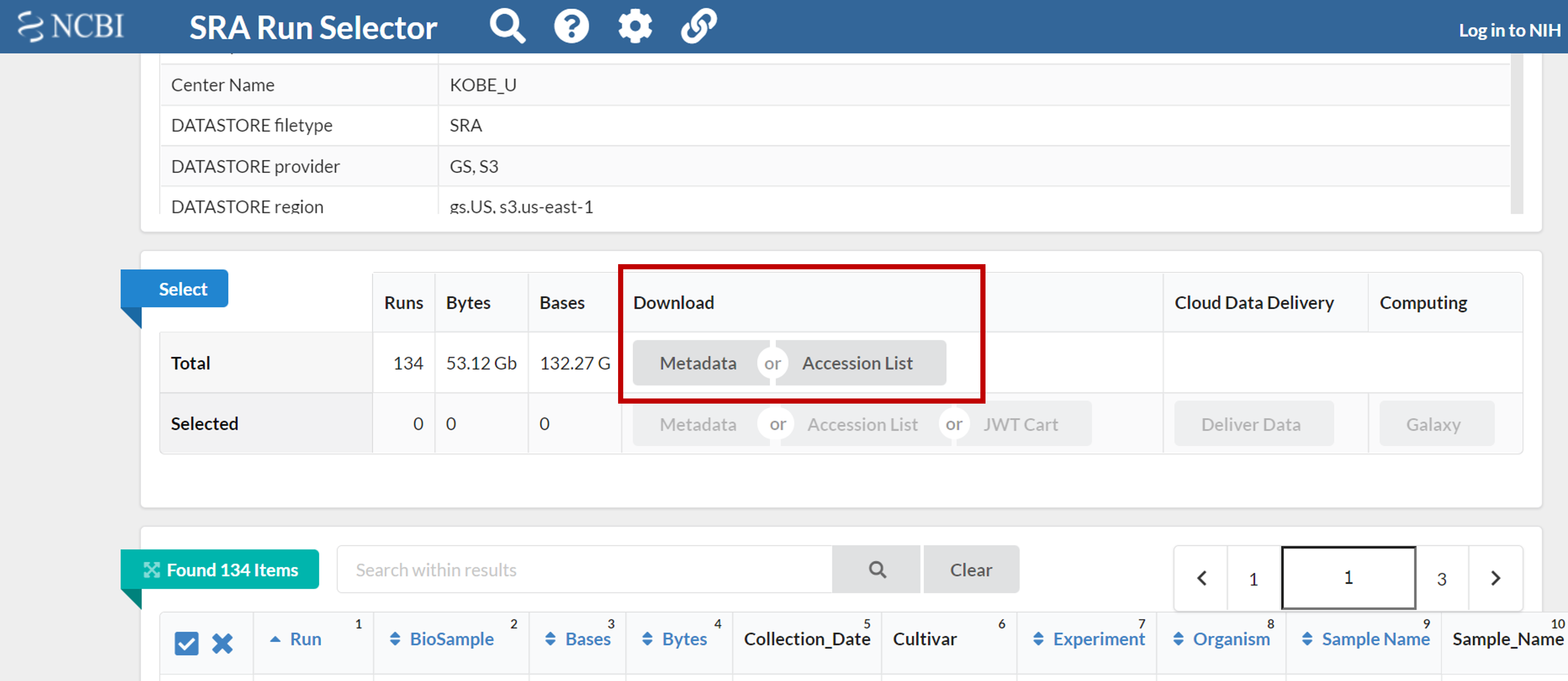
少し上にスクロールして、画面中央部にある「Accession List」というボタンをクリックすると、全てのSRA Run accession No.が記載されたtxtファイルがダウンロードできます。また、「Metadata」ボタンをクリックすることで、サンプル名を含む、シークエンスデータの詳細情報がコンマ区切りのtxtファイルでダウンロードできます。
これでFastqファイルのダウンロードに必要な情報が揃いました。
3. Fastqファイルのダウンロードにおススメのツール
SRA Toolkitという、NCBI公式のSequence Read Archivesのデータを扱うソフトウェア群が有名です。バージョン2.9.1から、fasterq-dumpというツールが使えるようになっています。
3.1 SRA Toolkitのインストール
上記のgithubサイトに詳細が記載されていますが、大まかには下記の通りです。コマンド例はUbuntu用をインストールする場合を記載しています。
3.1.1 SRA Toolkitのダウンロード
#Ubuntuの場合
wget --output-document sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
#CentOSの場合
wget --output-document sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz
#Mac OS Xの場合
curl --output sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-mac64.tar.gz3.1.2 SRA Toolkitの展開(解凍)
tar xvf sratoolkit.tar.gz3.1.3 PATHの設定
PATH(パス)とは、簡単にいうと「ファイルやフォルダの置いてある場所を示す情報」です(詳しくはこちら)。以下のように環境変数$PATHを設定しておくと、例えば「fasterq-dump」と打ち込むだけで目的のプログラムを走らせることができるようになります(これをしないと、/sratoolkit.2.11.2-ubuntu_linux64/bin/fasterq-dumpのようにいちいちフルパスを入力しなければならなくなるので面倒)。
#SRAToolkitをインストールしたディレクトリに入り、binに移動
cd sratoolkit.2.11.2-ubuntu_linux64/bin
pwd
#すると、fasterq-dumpがインストールされているディレクトリのPATHが表示される
/sratoolkit.2.11.2-ubuntu_linux64/bin
#PATHを通す
export PATH=$PATH:/sratoolkit.2.11.2-ubuntu_linux64/bin4. Fastqファイルのダウンロードの手順
4.1 基本の使い方
次のように入力すれば、Run accession No.毎に1ペアずつFastqファイルをダウンロードすることができます。
#SRRxxxxxxの部分には、ダウンロードしたいサンプルのaccession No.を入力
fasterq-dump -p SRRxxxxxx4.2 複数ファイルを一気にダウンロードしたい場合
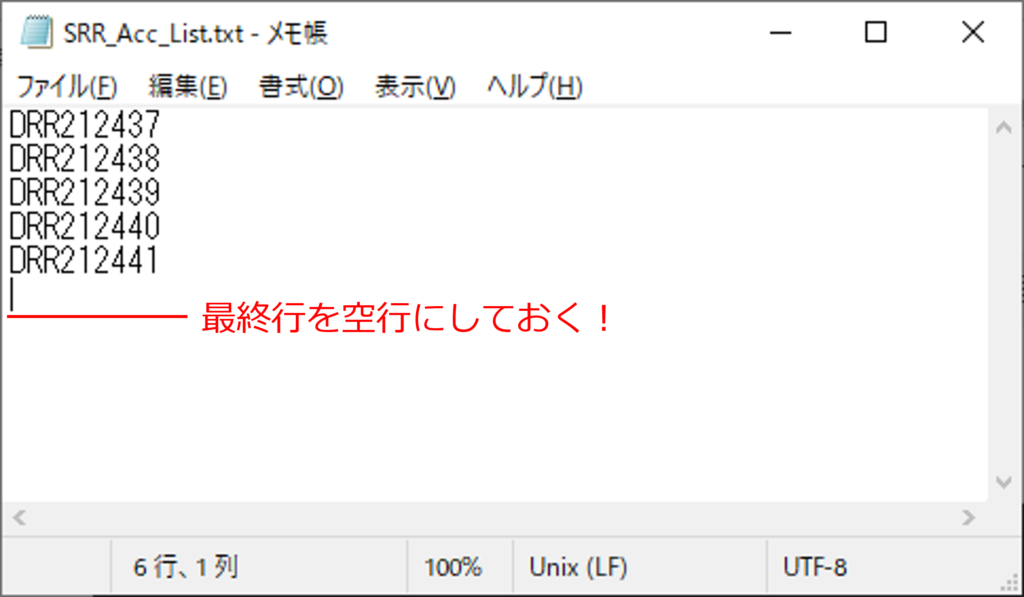
まず、ダウンロードしたいFastqファイルに紐づいているRun accession No.を下記のようにtxtファイルとして準備します(ファイル名は仮に「SRR_Acc_List.txt」としておきます)。この際、最終行を「空行」にしておくのがコツです。
このSRR_Acc_List.txtを適当なディレクトリに入れ、下記のようなコードを打つと、Fastqファイルのダウンロードが始まります。
while read line
do fasterq-dump -p $line
done < ./SRR_Acc_List.txt次のような画面が表示され、
join :|-------------------------------------------------- 100%
concat :|-------------------------------------------------- 100%
spots read : 2,838,879
reads read : 5,677,758
reads written : 5,677,758ls
DRR212437_1.fastq DRR212438_1.fastq DRR212439_1.fastq DRR212440_1.fastq DRR212441_1.fastq SRR_Acc_List.txt
DRR212437_2.fastq DRR212438_2.fastq DRR212439_2.fastq DRR212440_2.fastq DRR212441_2.fastqSRR_Acc_List.txtに対応するFastqファイルが一括でダウンロードできました!
Follow me!


コメント